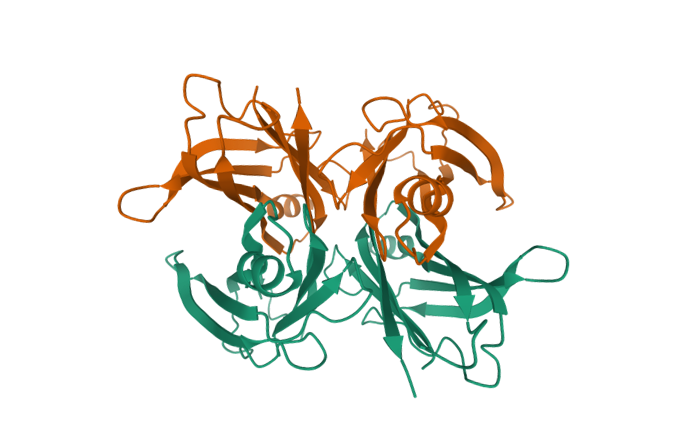ATTR Disease Overview
HCP Video: ATTR Disease Overview
- Amyloidosis Research Consortium, https://www.youtube.com/watch?v=lXZsWTUjV7Q
- Mayo Clinic. https://www.youtube.com/watch?v=RnWxzRD1NVk
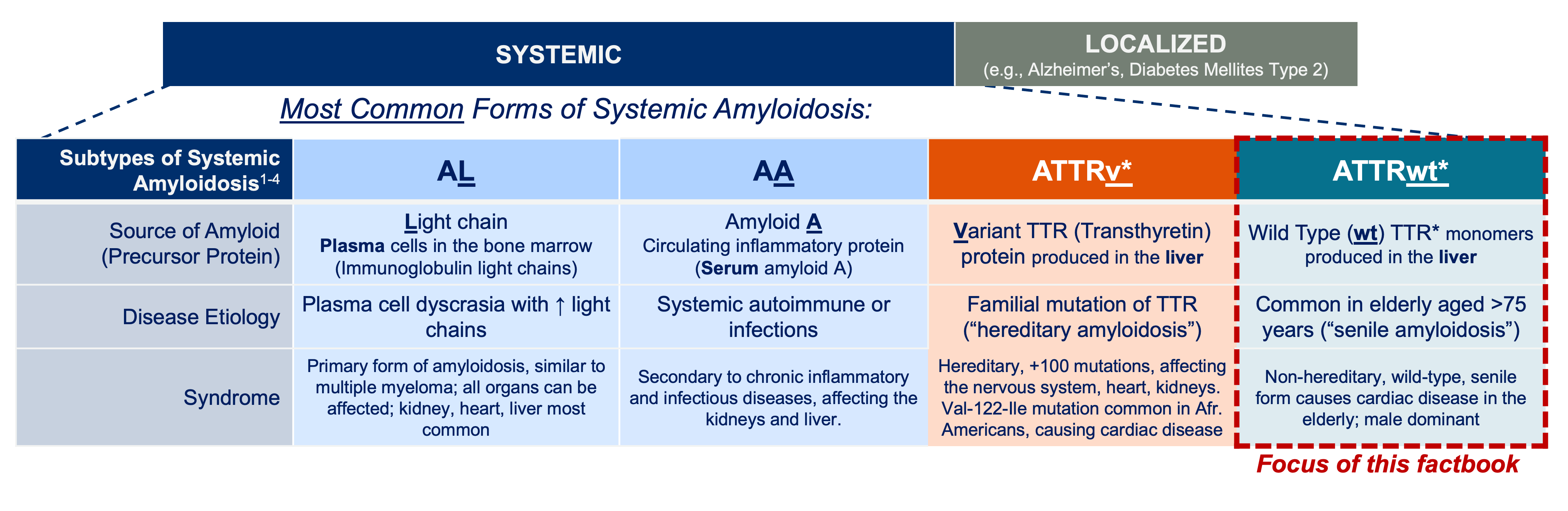
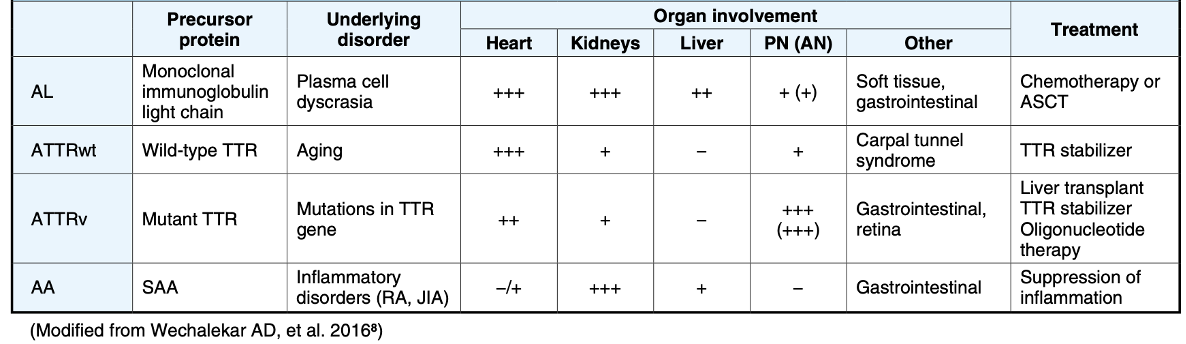
a. E.g., unlike AL Amyloidosis, AA Amyloidosis, ATTRv, and ATTRwt are non-neoplastic and will not benefit from chemotherapy [3]
b. Virtually all heredofamilial amyloidoses associated with nephropathic, neuropathic, or cardiopathic disease are dominantly inherited heterozygous disorders, and both the wild-type and mutant molecules can be identified in the amyloid deposits. In some instances (e.g., TTR, apolipoprotein A-I [ApoAI], Alzheimer APP, and prion protein [PRP]), both the wild-type and mutant molecules are able to form amyloid fibrils under different circumstances, with the wild-type protein implicated in aging-associated diseases. As an example, wild-type TTR; ApoAI; and the beta protein, A-beta, a cleavage product of APPs, may form deposits in association with organ-specific pathology in the aging heart, aorta, and brain, respectively. [6]
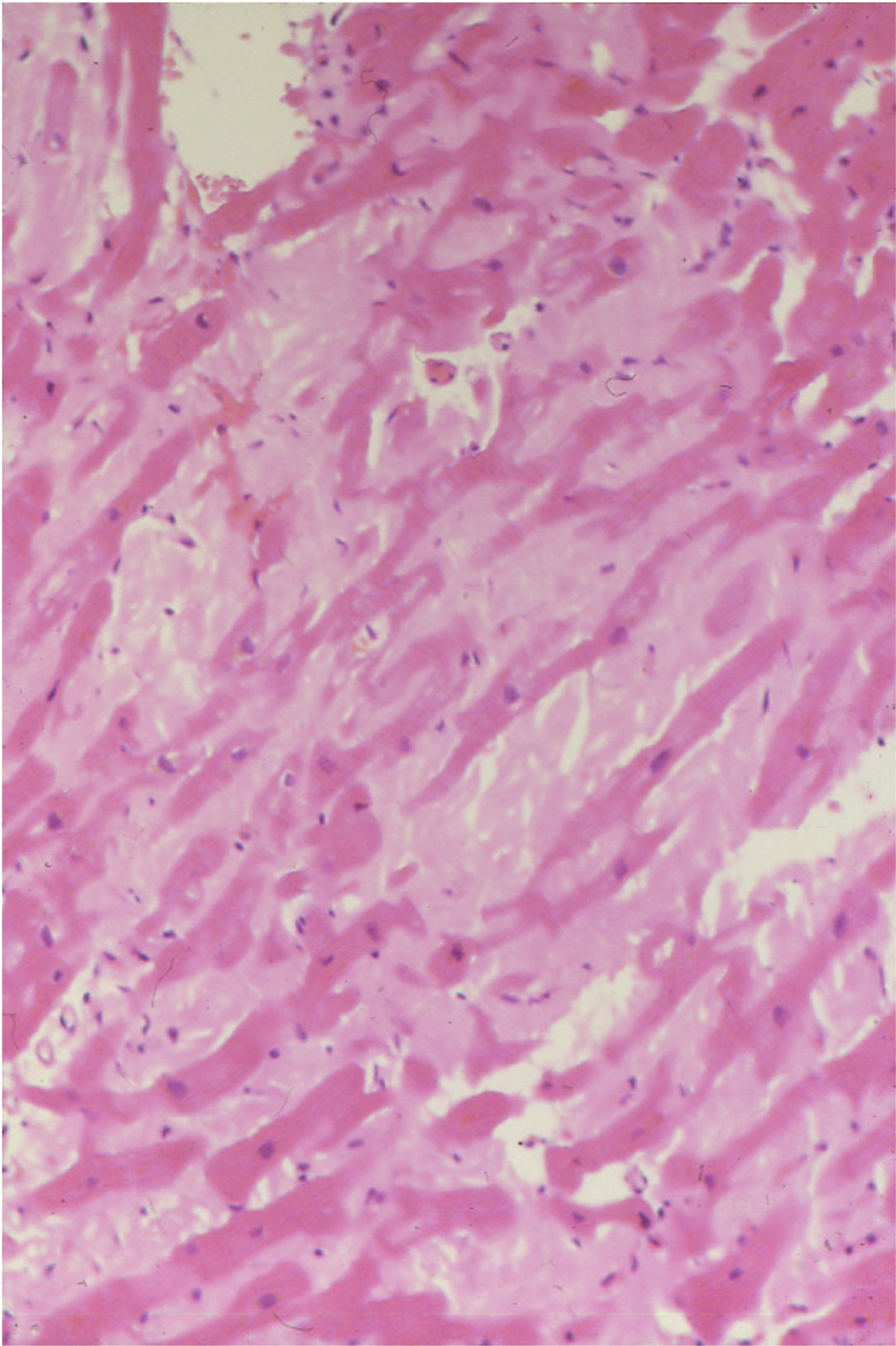
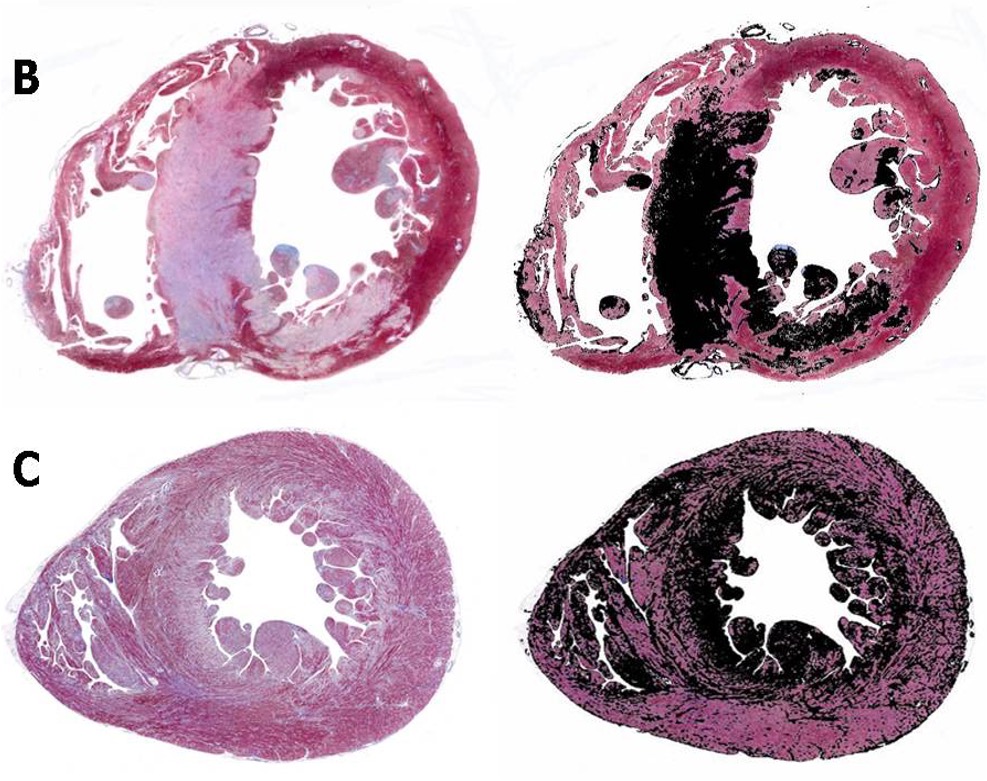
c. In the case of localized amyloidosis, the amyloid deposits are observed in the organ / tissue where the precursor protein is synthesized; whereas in systemic amyloidosis, the deposition of aggregates and fibrils occurs at locations different than the sites where the precursor protein is expressed. [4]
Glossary:
Glossary:
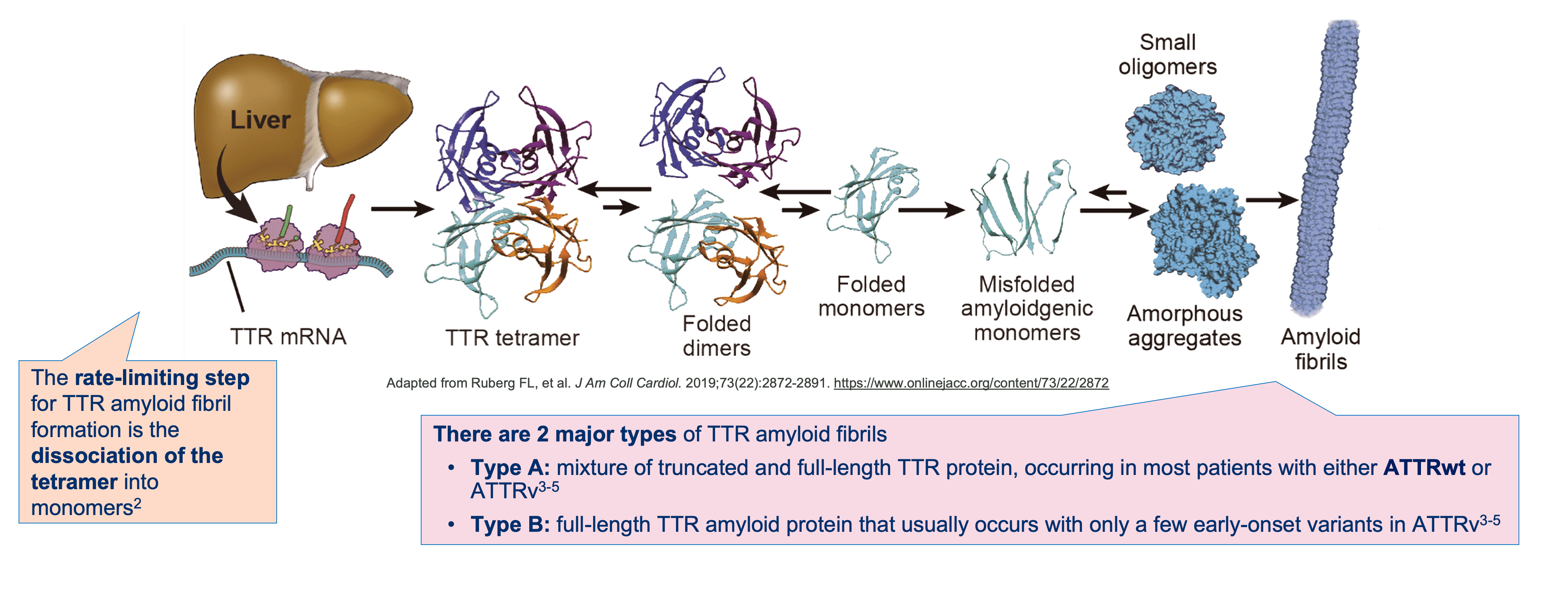
Glossary:
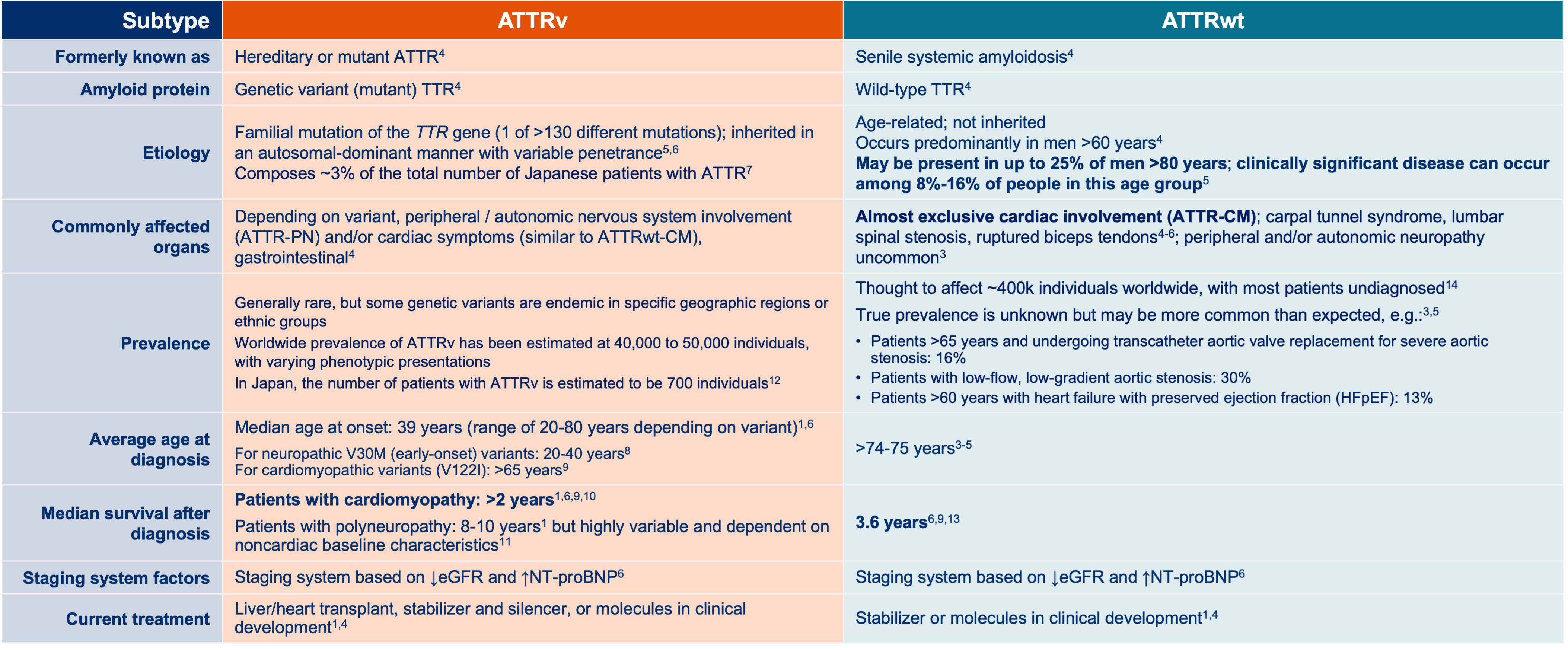
Glossary: