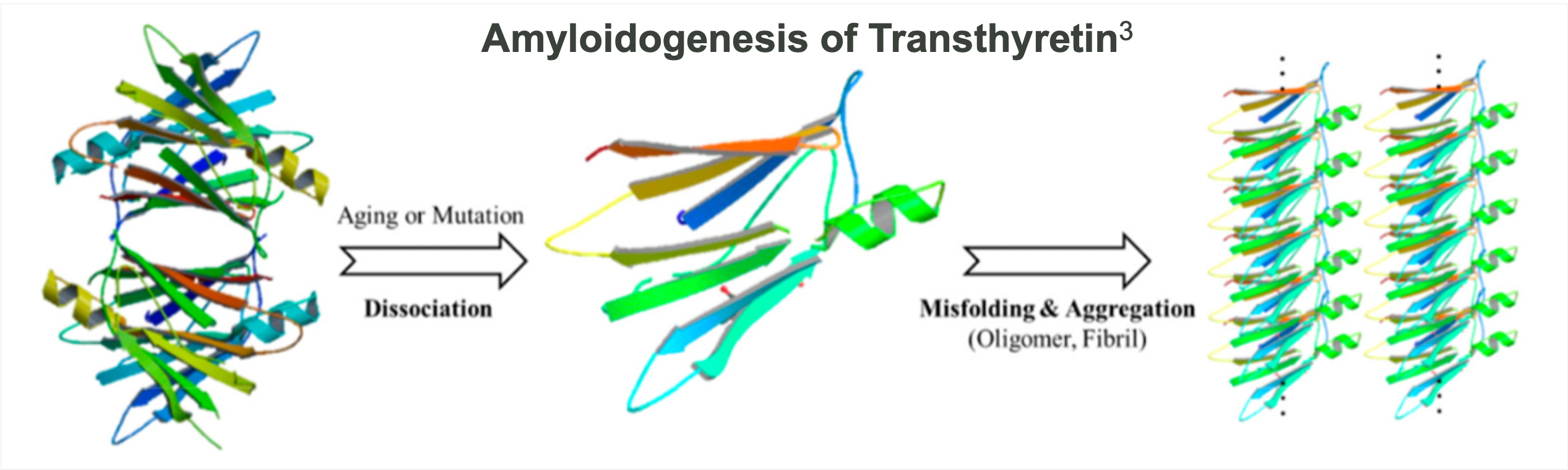
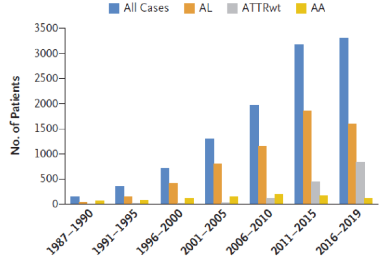
Abbreviated list of amyloid fibril proteins and their precursors in humans [1]
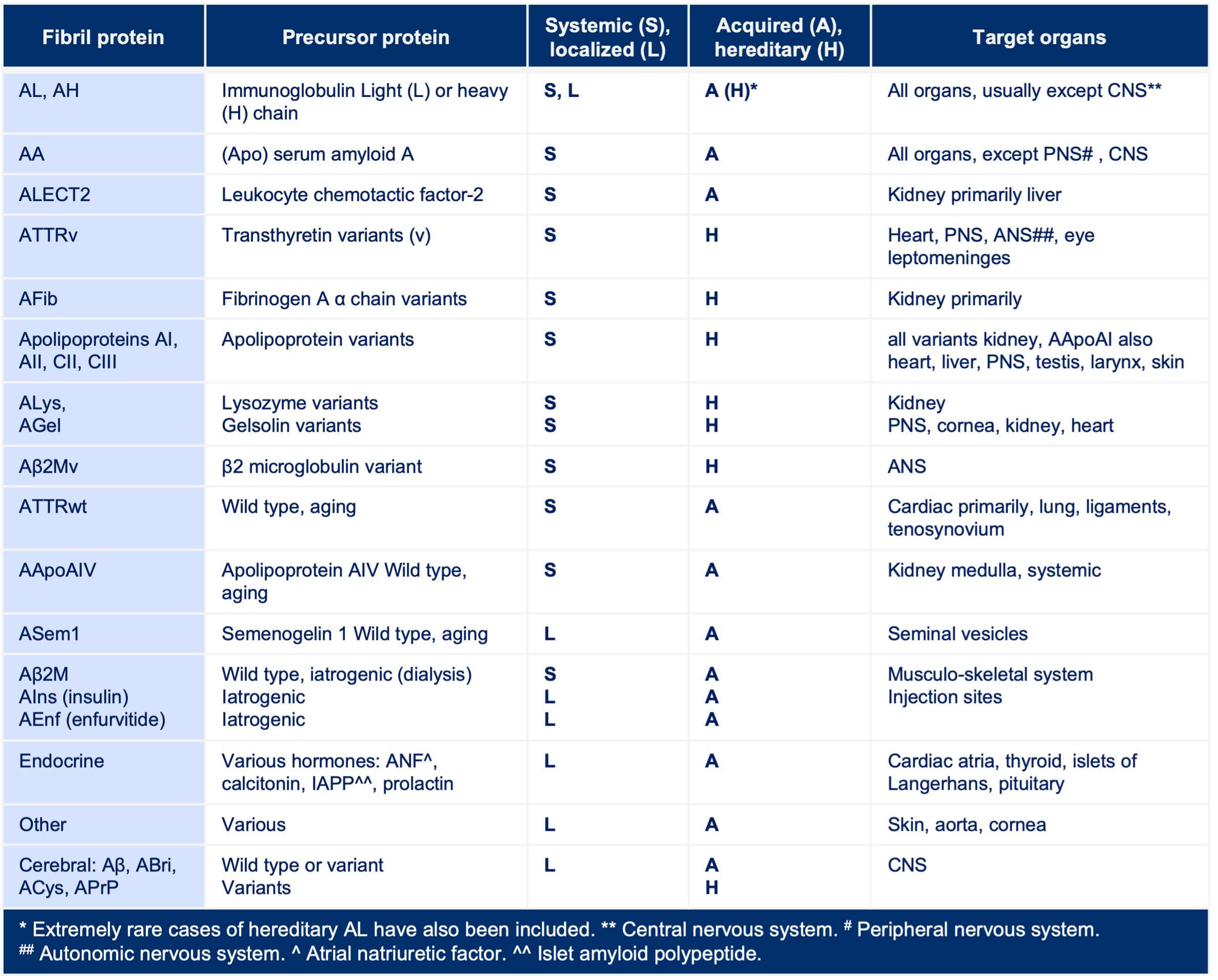
At least 38 different human protein precursors of amyloid fibrils are known—some are produced at the site of amyloid formation (localized), and some circulate in the blood to deposit in a variety of tissues and organs (systemic). [2]
Exclusively localized amyloid deposits have been associated with at least 19 protein types, while at least 14 protein types (and many more variants) appear to be consistently associated with systemic amyloidosis. [1]
Interestingly, however, at least 3 protein types (most notably AL/AH, amyloidosis derived from immunoglobulin light or heavy chain, respectively, amyloidosis derived from β2 microglobulin, Aβ2M), and ATTR can occur as either localized or systemic deposits. [1]
Exclusively localized amyloid deposits have been associated with at least 19 protein types, while at least 14 protein types (and many more variants) appear to be consistently associated with systemic amyloidosis. [1]
Interestingly, however, at least 3 protein types (most notably AL/AH, amyloidosis derived from immunoglobulin light or heavy chain, respectively, amyloidosis derived from β2 microglobulin, Aβ2M), and ATTR can occur as either localized or systemic deposits. [1]
Per the International Society of Amyloidosis (ISA) guidelines, in human medicine, the term amyloid is applied to mainly extracellular deposits of a fibrillary protein that are recognizable by their affinity for Congo red and their yellow-green birefringence under polarized light.
The current classification of amyloid in medical practice is based on the amyloid protein type. The amyloid is termed A (for amyloid) followed by an abbreviation of the protein type: AL (amyloid derived from immunoglobulin light chain), ATTR (amyloid derived from transthyretin), etc. [1]
The current classification of amyloid in medical practice is based on the amyloid protein type. The amyloid is termed A (for amyloid) followed by an abbreviation of the protein type: AL (amyloid derived from immunoglobulin light chain), ATTR (amyloid derived from transthyretin), etc. [1]
- Picken, MM. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020;143:322-334. doi: 10.1159/000506696.
- Gorevic, Peter. Aug. 10, 2021. Overview of amyloidosis. UptoDate. https://www.uptodate.com/contents/overview-of-amyloidosis?search=Symptomatic%20Transthyretin%20Amyloid%20Cardiomyopathy%20&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1.